Hematologic Disorders
Subtopic:
Coagulopathies
COAGULOPATHIES
Review of physiology blood clotting: Hemostatic Mechanism
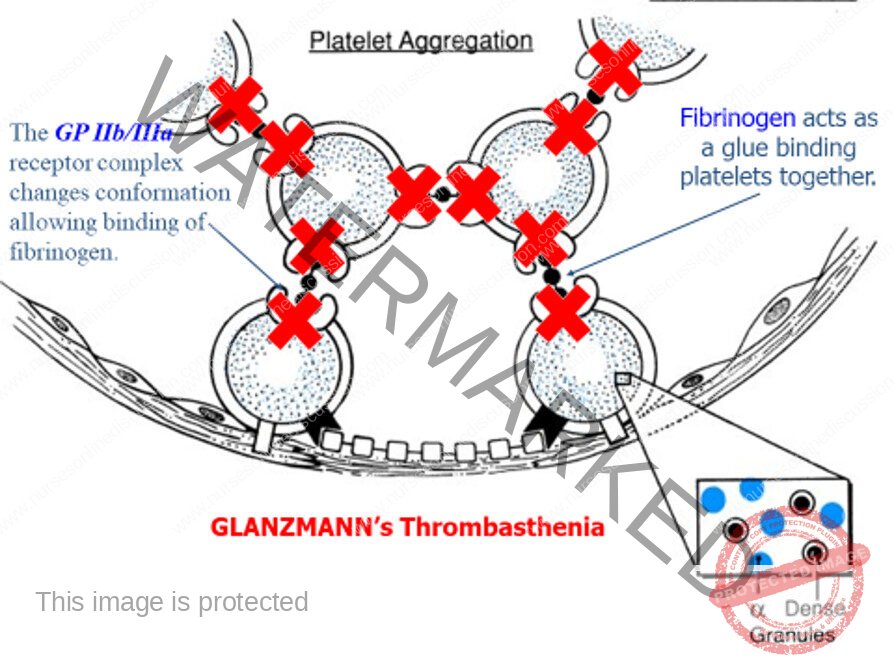
- Platelet adhesion
- Platelet aggregation
- Clot formation
- Clot stabilization
- Limitation of clotting to the site of injury by regulatory anticoagulants, and
- Re-establishment of vascular patency through fibrinolysis and vascular healing

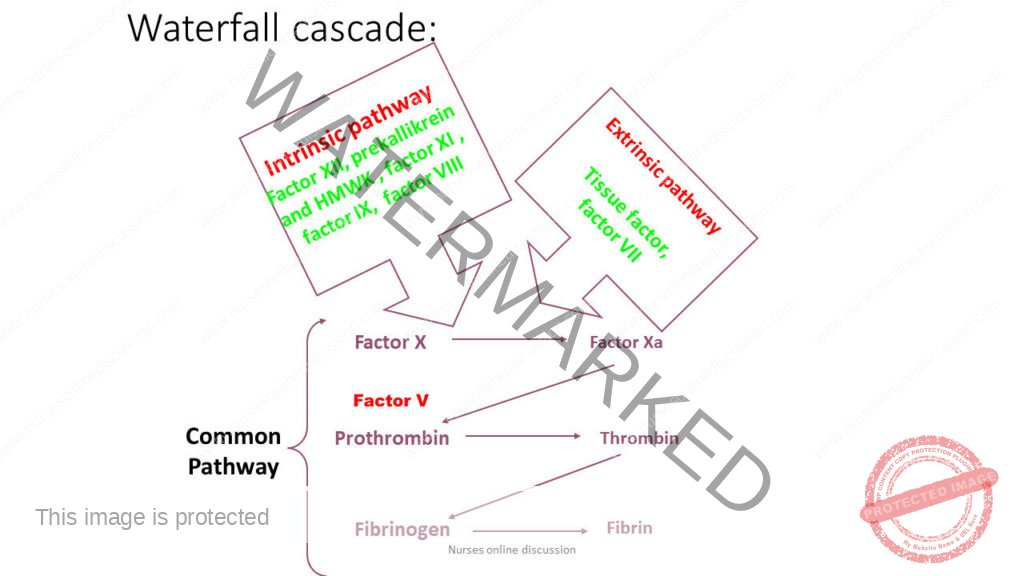
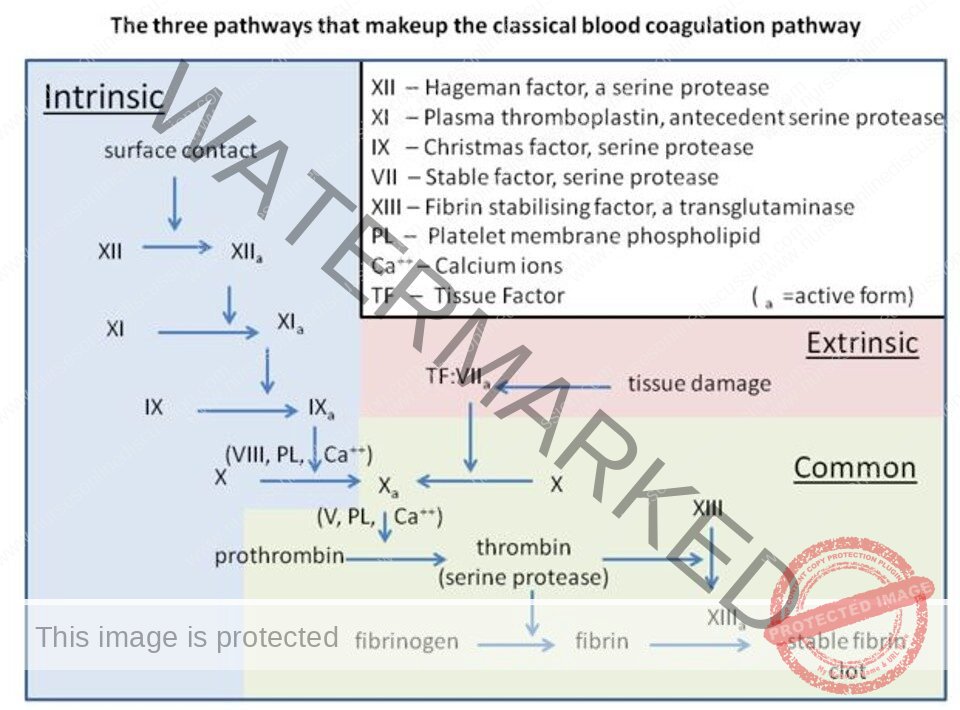
Clotting Factors
- Fibrinogen
Prothrombin
Labile factor, proaccelerin
Stable factor or proconvertin
Antihemophilic factor (AHF)
Christmas factor
Stuart-Power factor
Plasma thromboplastin antecedentHageman factor
Fibrin Stabilizing Factor

Definition of coagulopathies
- Coagulopathies are conditions that affect the blood’s clotting activity and result into either decreased coagulability states (hypocoagulability/bleeding disorders) or increased coagulability states (hypercoagulability/thrombocytosis).
Cont`
Inherited Coagulopathies
- Hemophilia A and B
- Von Willebrand disease
- Factor XIII deficiency
- Fibrinogen deficiency
Acquired Coagulopathies
- Vitamin K deficiency
- Warfarin therapy
- Liver disease
- Disseminated intravascular coagulation (DIC)
- Trauma-induced coagulopathy
Predisposing factors to coagulopathies
Hypercoagulability: Acquired factors
- Advanced age
- Immobilization
- Inflammation
- Pregnancy
- Oral contraceptive use
- Obesity
- DM
- Hormonal replacement therapy
- Cancer
- Antiphospholipid syndrome
Hypercoagulability: genetic/hereditary
- Protein C deficiency
- Protein S deficiency
- Antithrombin deficiency
- Factor V Leiden
- Prothrombin G2021OA/factor II mutation
Factors of primary bleeding disorder
- These predispose to reduced levels of platelets;
- Antiplatelet drugs; triagrelor, aspirin, prasugrel, clopidogrel etc.
- Disseminated intravascular coagulation (DIC).
- Bone marrow suppression; bone marrow aplasia/cancer/cancer therapy.
Factors of secondary bleeding disorder
- These predispose to reduced clotting factors or blocking their activity;
- Extensive liver disease/failure
- Vitamin K deficiency especially in neonates.
- Anti-coagulants such as Warfarin, heparin
Classification of coagulopathies
Coagulation disorders arise due to deficiency of one or more coagulation factor.
They are classified into two groups;
- Inherited (congenital) deficiency
- Haemophilia A
- Christmas disease (haemophilia B)
- Acquired deficiency
- Disseminated intravascular coagulation
- Vitamin K deficiency, e.g. obstructive jaundice, malabsorption
- Haemorrhagic diseases of the new born.
- Diseases of the liver
- Anticoagulant therapy
HEMOPHILIA
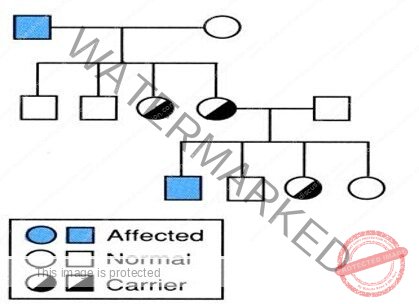
Often called ‘the disease of kings’ because it was carried by many members of Europe’s royal family. Queen Victoria of England was a carrier of Hemophilia
- Definition;
- It is an X-linked recessive disorder due to either;
- Factor VIII deficiency: Hemophilia A 80-85%
- OR
Factor IX deficiency: Hemophilia B
Aetiology;
- Mutations of the clotting factor gene
- Family H/O bleeding common,
– generally affects males on the maternal side
– 1/3 no family history – due to new mutations
- The disorder being transmitted as X-linked recessive manner, hence males suffer from the disease while the females act as carriers and transmit the disease their further children.
- Definition;
- It is an X-linked recessive disorder due to either;
- Factor VIII deficiency: Hemophilia A 80-85%
- OR
Factor IX deficiency: Hemophilia B
Aetiology;
- Mutations of the clotting factor gene
- Family H/O bleeding common,
– generally affects males on the maternal side
– 1/3 no family history – due to new mutations
- The disorder being transmitted as X-linked recessive manner, hence males suffer from the disease while the females act as carriers and transmit the disease their further children.
Types

Severity of Hemophilia is defined by measured level of clotting factor activity


CLINICAL MANIFESTATIONS
- Bleeding can happen anywhere in the body.
- Following an injury / surgery or rarely spontaneous



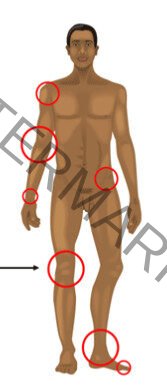
CLINICAL MANIFESTATIONS
Musculoskeletal bleeding
- Deep bleeding into joints and muscles
- Begin when child reaches toddler age.
- In toddlers ankle the most common site.
- Later knees and elbow become common sites
Hemophilic arthropathy
- Target joint
- Repeated bleeds

Other manifestations
- Intracranial haemorrhage
- Hematuria
- Traumatic bleeding
- Venipuncture

Carrier state and Genetic testing
Three approaches:
- Patient and family history
- Coagulation-based assays: unreliable
- DNA testing: GOLD standard
Hemophilia: Management
Issues to be considered
- Lifestyle modifications
- Available therapeutic options
- Inhibitors complicating Hemophilia
- Prophylactic factor therapy
- Transfusion transmitted infections
Hemophilia : Diagnosis
Screening tests
- Normal Prothrombin Time (PT) , Raised A partial Thromboplastin Time.(APTT)
- Mixing studies

- F VIII deficient plasma
- F IX deficient plasma
Definitive diagnosis specific factor VIII or IX by assays
Hemophilia: Management
Lifestyle modifications: Goal – Prevention of bleeding.
- Avoid drugs that affect platelet function -NSAIDs
- Paracetamol – safe for analgesia.
- Regular exercise to promote strong muscles, protect joints, and improve fitness.
- Avoid contact sports ; swimming and cycling encouraged.
- Recognize early signs of bleeding – a tingling sensation or “aura”.
- trained to seek treatment at this stage.
- Carry identification indicating the diagnosis, severity, and contact information.
Available pharmacological agents
- Factor concentrates
- Cryoprecipitate
- Fresh Frozen Plasma and Cryo-Poor Plasma
- Adjuvant Pharmacological Options
- Desmopressin (DDAVP)
- Tranexamic acid
- Epsilon aminocaproic acid (EACA)
Cryoprecipitate
– Prepared by slow thawing of FFP at 4°C for 10–24 hours.
– Contains – FVIII, vWF, fibrinogen, & FXIII (not FIX or XI).
– supernatant – cryo-poor plasma and contains other coagulation factors VII, IX, X, and XI.
– FVIII /bag of cryoppt is 60-100 units (avg-80 units) in a 30-40 ml vol.
-does not contain factor IX, so no use in Haemophilia B
Concerns :
– factor content of individual packs variable.
– not subjected to viral inactivation procedures
Fresh Frozen Plasma
- FFP can be used to treat both hemophilia A &B
- 1 U FFP contains about 160-250ml plasma with activity of ~80%.
- Rate and total dose limited by the risk of acute or chronic circulatory overload.
- How to use
- Thaw.
- Transfuse over how many minutes.
- Reusing after thawing
- Disadvantages:
- No viral inactivation
- F level >20-25% difficult to achieve
Desmopressin
- Only effective in mild hemophilia A – single i.v. infusion of 0.3 mg/kg expected to boost FVIII level 3-6 fold
- Ineffective in severe hemophilia A
- No value in hemophilia B – does not affect FIX levels
- Nasal spray available – useful for home treatment of minor bleeding problems.
Tranexamic acid / EACA
- Antifibrinolytic agent, competitively inhibits activation of plasminogen to plasmin.
- Valuable in controlling bleeding from mucosal surfaces (e.g., oral bleeding, epistaxis, menorrhagia)
- dental surgery
- eruption of teeth
- Tranexa dose for children – 25 mg/kg up to three times daily
– 500 mg tablet can be crushed, dissolved in water for topical use on bleeding mucosal lesions.
Management of Hemophilic arthropathy
- Analgesics, ice packs ( 5 minutes on, 10 minutes off, for as long as the joint feels hot), avoidance of weight bearing and immobilisation.
- Factor replacement- most important
- Physiotherapy
Prophylactic Therapy
- Administration of clotting factors at regular intervals to prevent bleeding
– Patients with clotting factor level > 1% seldom have
spontaneous bleeding
- 25-40 IU/kg of clotting factor concentrates
– 3 times/week for hemophilia A
– twice a week for hemophilia B
- Expensive but preservation of joint function & improved QOL
- Administered by subcutaneous access port of central line
Disseminated Intravascular coagulation (DIC)
- Definition; It is an acquired hemorrhagic disorder in which there is endothelial damage which initiates the coagulation cascade with the consumption of clotting factors and platelets leading to hemorrhage and microthrombosis at different sites.
- Causes; any condition resulting into endothelial damage
- Infections; gram negative septicemia, meningococcal septicemia, certain viral infections, malaria, snake bites
- Liberation/release of tissue factor (thromboplastin); abruptio placentae, retained dead placenta, amniotic fluid embolism, pre-eclampsia, massive trauma, malignancies
- Wide spread endothelia damage; severe burns, haemolytic uremic syndrome.
- Clinical features; bleeding from different sites; skin, mucous membrane, surgical incision sites, venepuncture, catheter sites etc. bleeding from more than two sties at the same time suggests DIC, gangrenous toes and fingers due to microthrombi.
- Diagnosis confirmed by;
- Low platelet count and fibrinogen level
- Prolonged prothrombin time and index (PT &PI) and partial thromboplastin time (PTTK)
- Presence of fibrinogen degradation products.
- Rx;-identifying and treating underlying cause
Von willebrand disease.
- Von Willebrand disease is a common, inherited, genetically and clinically heterogeneous hemorrhagic disorder caused by a deficiency or dysfunction of the protein termed von Willebrand factor.
- Consequently, defective factor interaction between platelets and the vessel wall impairs primary hemostasis.
- Von Willebrand factor (VWF) is a blood glycoprotein that plays a crucial role in hemostasis.
- Hemostasis is the body’s natural response to an injury that causes bleeding. It’s a complex process that stops bleeding and allows the body to start repairs
Clinical features vWD;
- Easy bruising
- Epistaxis
- Bleeding from the gum
- Hemorrhage after laceration/surgery
- Menorrhagia
- GI bleeding
- Excessive postpartum bleeding
- Massive soft tissue hemorrhage and bleeding into joints.
Diagnosis
- Prolonged bleeding time
- Slightly prolonged partially thromboplastin time
- Absent or low factor viii
- Absent or low factor viii related antigens
- Low factor viii activity
- Normal platelet count and clot retraction.
Haemorrhagic disease of the new borne
- This usually manifests during the first week of life in form of epistaxis (nose bleeding) or melana (blood in stool).
- The cause is hypoprothrombinemia (low prothrombin level) brought about by a shortage of Vitamin K received from the mother combined with inability of the neonate to synthesize Vitamin K itself in the first week of life due to absence of bacterial flora in the infant’s GIT.
- Prematurity predisposes to this condition hence premature infants must be given Vitamin K after birth as a routine. And also for all babies at birth as a prophylaxis.
Anticoagulant induces haemorrhage
- Anti-coagulants are used to treatment thrombosis and embolism, especially venous thrombosis. These drugs prolong bleeding time (heparin) and clotting time (oral anticoagulants) by affecting the intrinsic and extrinsic pathway, thus prevention of clot formation.
- Nursing care for a patient on anticoagulant
- Monitoring the clotting profiles; (BT, CT, PTTK).
- Daily routine gross and microscopic examination of the urine for RBCS
- Observe for signs of bleeding; rapid pulse, hypotension or new haemorrhagic spots and report immediately.
- Report any suspected allergic reaction immediately
- Avoid trauma or IM injections when patient is on anti-coagulants.
- Have Vitamin K or protamine at hand to treat any bleeding.
- Rx; -anticoagulant is stopped immediately
- -give appropriate anti-dote e.g. protamine for heparin and vitamin K for oral anticoagulants.
- -transfusion with fresh frozen plasma that contain all the clotting factors.
We are a supportive platform dedicated to empowering student nurses and midwives through quality educational resources, career guidance, and a vibrant community. Join us to connect, learn, and grow in your healthcare journey
Quick Links
Our Courses
Legal / Policies
Get in Touch
(+256) 790 036 252
(+256) 748 324 644
Info@nursesonlinediscussion.com
Kampala ,Uganda
© 2026 Nurses online discussion. All Rights Reserved