Paediatrics II
Congenital abnormalities (GIT, genito urinary, cardiovascular, CNS, musculo–skeletal)
Table of Contents
Congenital Abnormalities (GIT, Genito Urinary, Cardiovascular, CNS, Musculo-Skeletal)
General Definition
Congenital abnormalities, also known as congenital disorders or birth defects, are structural or functional anomalies that occur during intrauterine life.
These conditions can be identified prenatally, at birth, or sometimes may only be detected later in infancy. Some disorders however may remain unrecognized.
It is any defect in form, structure or function or it is a malformation of the fetus which is present at birth and can be detected on examination of the newborn after 1 hour of birth.
Causes of Congenital Abnormalities
Why some fetal tissues develop abnormally is not fully known but the following factors are thought to be responsible.
Predisposing Factors
- Maternal age: The risk of having a baby with an abnormality increases with age. Women above the age of 35 are more at risk of having babies with Down’s syndrome.
- Paternal age: Increased paternal age has been linked to dwarfism, although men can father children up to 90 years of age.
- Nutritional factors: Anoxia or hypoxia in utero can seriously affect growing brain cells if the mother’s blood is deficient in oxygen. This can occur in conditions like serious cardiac diseases, general anesthesia, and partial placental separation, as may occur in threatened abortion, and other perinatal complications (e.g., hypoxia, asphyxia, trauma).
Others can be categorized as:
- Chromosome and gene abnormalities: Each human human cell carries a print for reproduction in the form of 44 chromosomes (autosomes) and two sex chromosomes. Each chromosome comprises genes. Should any fault occur in the formation of gametes, the defect is transmitted via the genes in the ovum or spermatozoa, resulting in abnormalities like Down’s syndrome, anencephaly, cleft palate and harelip or cleft lip.
- Teratogenic causes: A teratogen is any agent that raises and affects the well-being of the fetus. Factors influencing the effects produced by the teratogen include gestational age of the embryo at the time of exposure, length of exposure, and toxicity of exposure. Examples include: a. Large concentrations of Vitamin A. b. Anticonvulsants. c. Tetracycline. d. Streptomycin. e. Thalidomide. f. Substance abuse: use of heroin, alcohol, nicotine from active or passive smoking.
- Environmental factors: a. Physical and chemical agents. b. Radiation. c. Diagnostic abdominal radiology x-rays (should be avoided during the first 30 weeks of pregnancy where growth and development of the fetus can be affected). d. Chemicals such as pesticides. e. Infectious agents, e.g., Rubella occurring during the first 12 weeks of pregnancy is a notable example. f. Cytomegalovirus.
- Intrauterine adverse effects during pregnancy, especially in the first trimester: These include drugs like Thalidomide, Streptomycin, and Tetracycline.
- Infections: Viruses such as HIV and TORCHES: a. TO – Toxoplasmosis. b. R – Rubella. c. C – Cytomegalovirus. d. HE – Herpes/Histoplasmosis. e. S – Syphilis.
- Maternal diseases: Conditions like hypertension, cardiovascular diseases, diabetes mellitus, and kidney disease.
- Abnormalities of the placenta and umbilical cord: These can lead to oxygen and nutrient deprivation, causing abnormalities of the fetus. Abnormal implantation of the ovum is also a contributing factor.
- Abnormal implantation of the ovum.
These abnormalities occur due to various reasons:
- Failure of tissues to engage: This happens when certain body tissues don’t connect properly during development. It could be like puzzle pieces not fitting together correctly. This can lead to problems in how the body functions or looks.
- Failure of an organ to form (Agenesis): Sometimes, an organ doesn’t develop at all. It’s like a missing piece in the body’s puzzle. For example, if a baby is born without a kidney, it’s called renal agenesis.
- Failure of canalization: Canalization is like the process of building a tunnel. Sometimes, parts of the body that should have a clear path, like the biliary tract (a pathway for bile in the liver) or the anus, don’t develop properly and become blocked. This can cause health issues, such as problems with digestion or waste removal.
- Failure of organs to separate: Sometimes, organs that should be separate end up joined together. It’s like two things sticking together when they shouldn’t, such as fingers fused together (syndactyly) or a kidney forming a horseshoe shape instead of being separate.
- Pressure on fetal limbs: When there’s pressure on the baby’s developing limbs in the womb, it can cause them to form incorrectly. This might lead to conditions like clubfoot, where the foot is twisted or turned inward.
- Failure to rotate: As the baby grows inside the womb, it needs to move and rotate to develop properly. If there’s not enough space for this movement, or if something restricts the baby’s movement, it might not rotate correctly. This can affect how the baby’s body forms, leading to problems like misaligned organs or limbs.
GIT (Gastrointestinal Tract) Abnormalities
Ankyloglossia (Tongue Tie)
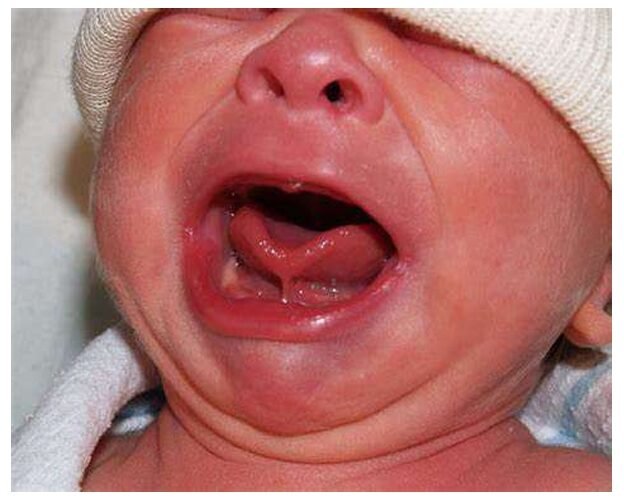
Ankyloglossia, commonly known as tongue tie, refers to an abnormal restriction of the tongue caused by a tight frenulum, the membrane attached to the lower anterior tip of the tongue (Kelley et al, 2008).
The exact cause of ankyloglossia isn’t fully understood, but it’s considered a congenital condition, meaning it’s present at birth. It’s believed to be primarily due to a failure of the lingual frenulum to fully separate during fetal development.
Thyroglossal Cyst
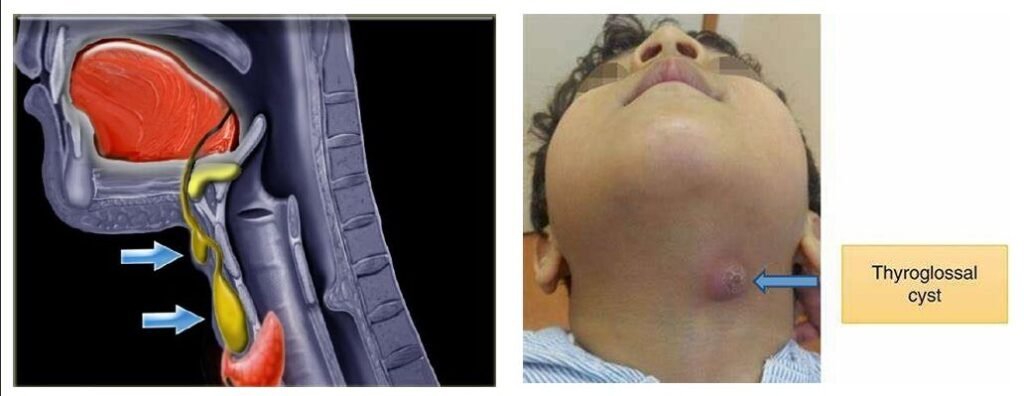
A thyroglossal cyst originates from an embryonic defect, resulting in the formation of a cyst at the base of the tongue.
A thyroglossal cyst is a benign (non-cancerous) mass that forms in the neck along the path of the thyroglossal duct.
This duct is a structure present during fetal development that connects the thyroid gland to the base of the tongue. Normally, this duct disappears before birth; however, if a portion of the duct persists, a cyst can develop.
Cleft Lip and Palate
Cleft lip and cleft palate are congenital (present at birth) craniofacial anomalies resulting from incomplete fusion of the embryonic facial tissues during the first trimester of pregnancy.
They are separate conditions, but often occur together.
A cleft lip is a split or opening in the upper lip, while a cleft palate is a split or opening in the roof of the mouth (palate).
A cleft palate is a congenital condition characterized by a split or opening in the roof of the mouth, resulting from incomplete fusion of the palatal shelves during embryonic development. This failure of fusion may occur during the first trimester of pregnancy, leading to the formation of a gap between the oral and nasal cavities (Cleft Lip and Palate Association, n.d.).
Tracheoesophageal Atresia and Fistula
Oesophageal atresia (OA) and tracheo-oesophageal fistula (TOF) are congenital anomalies of the oesophagus (food pipe) and trachea (windpipe) that occur during fetal development.
OA is the incomplete development of the oesophagus resulting in a gap or complete separation in the oesophagus, while TOF is an abnormal connection between the oesophagus and trachea.
During the development of a baby in the womb, around 4 to 8 weeks after conception, the laryngotracheal groove forms into the larynx, trachea (windpipe), and the early stages of lung tissue. At the same time, the esophagus, which is the tube that carries food from the mouth to the stomach, begins to take shape next to these structures.
Sometimes, certain factors or substances in the environment can disrupt this process, preventing the trachea and esophagus from separating properly. This can lead to various abnormalities, one of which is oesophageal atresia.
Omphalocele
Omphalocele is a congenital abdominal wall defect in which the intestines, liver, or other abdominal organs protrude through the umbilicus (belly button) into a sac.
This sac is covered by a thin membrane composed of amnion and peritoneum, making the organs visible. Unlike gastroschisis (another abdominal wall defect), the omphalocele is contained within this membrane. The herniated organs are usually the intestines but may include stomach and liver.
When the defect is less than 4 cm it is termed a hernia of the umbilical cord; when greater than 10cm, it is a true omphalocele.
This condition occurs because at approximately 6 to 8 weeks of intrauterine life, the fetal abdominal contents grow faster than the fetal abdomen. The occurrence is associated with chromosomal aberration (deviation) (Louik et al.,2007).
The exact cause is unclear, but it likely involves a failure of the abdominal wall to close properly during fetal development.
Gastroschisis
Gastroschisis is a congenital abdominal wall defect characterized by the extrusion of abdominal contents (intestines, and sometimes other organs) through a defect in the abdominal wall.
Unlike omphalocele, the defect is usually located to the right of the umbilicus and is not covered by a sac; the exposed organs are directly in contact with amniotic fluid. This exposure leads to significant inflammation and damage to the intestines.
Gastroschisis occurs due to a failure of the abdominal wall to close properly during fetal development, often unrelated to other congenital anomalies.
Intestinal Obstruction
Intestinal obstruction occurs when there is a partial or complete blockage of the intestines, leading to a disruption in the normal flow of intestinal contents.
Intestinal obstruction in newborns refers to a blockage of the intestinal tract that prevents the normal passage of stool. This can occur anywhere along the gastrointestinal (GI) tract, from the stomach to the anus, and is a serious condition requiring prompt medical attention.
Intestinal obstruction in newborns can result from congenital anomalies such as malrotation, volvulus, or atresia, as well as acquired conditions like intussusception or meconium ileus.
Meconium Plug Syndrome
Meconium plug syndrome, also known as meconium ileus equivalent, is a condition characterized by the failure of meconium to pass through the distal colon, resulting in obstruction.
A Meconium plug is an extremely hard portion of Meconium that has completely blocked the intestinal lumen, causing bowel obstruction.
Meconium plug syndrome occurs when thickened meconium forms a plug in the colon, leading to obstruction. It is often associated with cystic fibrosis.
Imperforate Anus
Imperforate anus is a congenital anomaly where the anus doesn’t form properly during fetal development. This means the rectum doesn’t connect to the outside of the body, preventing the passage of stool.
Imperforate anus is stricture of the anus (Vick et al., 2007). During the 7th week of intrauterine life. The upper bowel elongates to pouch and combine with a pouch invigilating from the perineum. There may be no rectum or Anus OR A rectum may be there but no opening OR The Anal sphincter may be enclosed by a membrane.
Imperforate anus occurs due to abnormal development of the fetal hindgut during embryogenesis.
Pyloric Stenosis
Pyloric stenosis is a condition affecting infants where the pylorus, the muscular valve between the stomach and the small intestine, becomes thickened and narrowed.
This narrowing obstructs the flow of food from the stomach to the duodenum, leading to forceful vomiting and dehydration.
The exact cause of pyloric stenosis is not fully understood, but it is believed to involve both genetic and environmental factors. There may be a familial predisposition, and certain factors such as erythromycin use during pregnancy have been implicated.
Genito Urinary Abnormalities
(Note: The document does not explicitly detail genito urinary abnormalities, but Imperforate Anus often has associated genito urinary anomalies, such as fistulas between rectum and urethra/vagina in high-type cases.)
Cardiovascular Abnormalities
Atrial Septal Defect (ASD)
Abnormal opening between the atria.
- Clinical Features: Often asymptomatic; may present with a systolic murmur, fatigue, shortness of breath (especially with exertion), and recurrent respiratory infections.
Atrioventricular Canal Defect
A combination of ASD and VSD with abnormal atrioventricular valves.
- Clinical Features: Similar to ASD and VSD but usually more severe; often associated with other congenital anomalies (e.g., Down syndrome).
Bicuspid Aortic Valve
Narrowing of the aortic valve.
- Clinical Features: May be asymptomatic; can present with a systolic ejection murmur, chest pain (angina), syncope (fainting), and signs of left ventricular hypertrophy.
Coarctation of the Aorta
Narrowing of the aorta.
- Clinical Features: High blood pressure in the upper extremities and low blood pressure in the lower extremities; may present with headaches, dizziness, nosebleeds, leg pain, and weak or absent femoral pulses.
Congenital Mitral Valve Anomalies
Narrowing of the pulmonary valve.
- Clinical Features: May be asymptomatic; can present with a systolic ejection murmur, right ventricular hypertrophy, and varying degrees of cyanosis if severe.
CNS (Central Nervous System) Abnormalities
Hydrocephalus
Hydrocephalus is a condition where there’s an abnormal buildup of cerebrospinal fluid (CSF) in the brain.
This fluid normally cushions the brain and spinal cord, but when it can’t drain properly, it puts pressure on the brain. This can cause the head to swell and lead to symptoms like headaches, nausea, vomiting, and changes in vision. In babies, whose skulls are still forming, it can cause the head to enlarge rapidly.
Convulsions in Infants and Young Children
Convulsions, also known as seizures, are involuntary spasmodic contractions of the skeletal muscles due to abnormal electrical activity in the brain.
They represent a malfunction of the brain’s electrical system and are a common neurological presentation in children, particularly during the first two years of life.
The underlying causes are diverse and necessitate a thorough investigation.
Musculo-Skeletal Abnormalities
Amelia and Ectromelia
- Amelia: The complete absence of one or more limbs. Thalidomide exposure during pregnancy is a known cause, but other genetic and environmental factors can also contribute.
- Ectromelia: The partial absence of one or more limbs, a more common occurrence than amelia. The severity can vary widely, ranging from minor limb shortening to significant malformations. Etiology is often multifactorial and may involve genetic factors, teratogens, and vascular disruptions during development.
Polydactyly (Extra Digits)
The presence of more than the usual number of fingers or toes. Diagnosis is typically straightforward through visual inspection. Management depends on the digit’s structure:
- Skin-only attachment: These extra digits can often be simply ligated (tied off) with a sterile thread at birth.
- Digit with bone and firm attachment: Surgical removal is usually necessary and should be performed by a surgeon specializing in pediatric orthopedics or hand surgery. The timing of surgery is determined by the surgeon based on the child’s age and development.
Syndactyly (Webbing of Digits)
Fusion of two or more fingers or toes. The degree of fusion varies. Surgical separation is typically performed to improve function and aesthetics, particularly if the digits are naturally separated but connected by skin or soft tissue. The timing of surgery is determined by the surgeon based on the child’s age and development.
Talipes (Clubfoot)
An abnormal positioning of the foot, often involving inversion (inward bending) and plantarflexion (pointing downwards). It’s frequently caused by intrauterine pressure, such as in multiple pregnancies (twins, triplets) or pregnancies with oligohydramnios (reduced amniotic fluid). Muscle tendon contractures contribute to the deformity.
- Talipes Equinovarus: The most common type, characterized by inward bending (inversion) of the foot and plantarflexion at the ankle.
- Talipes Calcaneovalgus: The foot is turned outward (eversion) with dorsiflexion (upward bending) of the ankle.
Achondroplasia (Chondrodystrophy)
A genetic disorder affecting bone growth, inherited as an autosomal dominant trait. It primarily disrupts cartilage production, particularly in the epiphyseal plates (growth plates) of long bones.
Congenital Dislocation of the Hip (CDH)
The hip joint is dislocated at birth. The cause is often laxity (looseness) of the hip joint capsule, potentially influenced by hormonal factors during pregnancy (e.g., progesterone).
Congenital Heart Disease (CHD)
Congenital heart disease encompasses a wide range of structural abnormalities of the heart present at birth.
These defects can affect the heart’s chambers, valves, great vessels (aorta and pulmonary artery), or the connections between them. These anomalies interfere with the normal flow of blood through the heart, leading to varying degrees of oxygen deficiency in the body.
CHD is a significant cause of morbidity and mortality in newborns and infants. The severity of CHD varies greatly; some conditions are mild and may not require intervention, while others are life-threatening and require immediate medical attention. The precise defect(s) present significantly impacts the clinical presentation and necessary management.
Aetiology of Congenital Heart Disease
The exact cause of most CHDs remains unknown, but several factors are believed to contribute:
- Genetic factors: Chromosomal abnormalities (e.g., Down syndrome, Trisomy 18), single-gene mutations, and familial predisposition play significant roles.
- Environmental factors: Maternal infections (rubella, cytomegalovirus), exposure to certain medications (e.g., lithium, thalidomide) during pregnancy, maternal diabetes, and alcohol consumption can increase the risk.
- Multifactorial inheritance: A combination of genetic and environmental factors likely contributes to many CHDs.
General Assessment/Investigations
The assessment and investigations will depend on the suspected CHD. Common methods include:
- History: Detailed prenatal, perinatal, and postnatal history is crucial (e.g., maternal illnesses, family history of heart defects, cyanosis, feeding difficulties).
- Physical examination:
- Auscultation (listening to the heart sounds for murmurs, extra heart sounds).
- Assessment of pulses (strength and equality).
- Observation for cyanosis (bluish discoloration of the skin).
- Assessment of respiratory rate and effort, and evaluation of growth and development.
- Electrocardiogram (ECG): Assesses the heart’s electrical activity, providing clues about the location and nature of the defect.
- Chest X-ray: Provides information about heart size, pulmonary blood flow, and lung fields.
- Echocardiogram: A non-invasive ultrasound of the heart that provides detailed images of the heart’s structures and blood flow patterns; considered the gold standard for diagnosing CHD.
- Cardiac catheterization: A more invasive procedure involving inserting a catheter into the heart to obtain blood samples, measure pressures, and perform interventions.
Types of Congenital Heart Defects
- Acyanotic Defects (Increased Pulmonary Blood Flow): These defects result in increased blood flow to the lungs.
- Atrial Septal Defect (ASD): Abnormal opening between the atria.
- Clinical Features: Often asymptomatic; may present with a systolic murmur, fatigue, shortness of breath (especially with exertion), and recurrent respiratory infections.
- Ventricular Septal Defect (VSD): Abnormal opening between the ventricles.
- Clinical Features: Often asymptomatic, especially small VSDs; may present with a loud, harsh murmur, poor weight gain, failure to thrive, recurrent respiratory infections, and heart failure signs (tachypnea, tachycardia).
- Patent Ductus Arteriosus (PDA): Failure of the ductus arteriosus (a fetal blood vessel) to close after birth.
- Clinical Features: May be asymptomatic, or present with a continuous “machinery” murmur, bounding peripheral pulses, and symptoms of heart failure (especially in larger PDAs).
- Atrioventricular Canal Defect (AV Canal): A combination of ASD and VSD with abnormal atrioventricular valves.
- Clinical Features: Similar to ASD and VSD but usually more severe; often associated with other congenital anomalies (e.g., Down syndrome).
- Atrial Septal Defect (ASD): Abnormal opening between the atria.
- Acyanotic Defects (Obstructive Lesions): These defects obstruct blood flow.
- Pulmonary Stenosis (PS): Narrowing of the pulmonary valve.
- Clinical Features: May be asymptomatic; can present with a systolic ejection murmur, right ventricular hypertrophy, and varying degrees of cyanosis if severe.
- Aortic Stenosis (AS): Narrowing of the aortic valve.
- Clinical Features: May be asymptomatic; can present with a systolic ejection murmur, chest pain (angina), syncope (fainting), and signs of left ventricular hypertrophy.
- Coarctation of the Aorta: Narrowing of the aorta.
- Clinical Features: High blood pressure in the upper extremities and low blood pressure in the lower extremities; may present with headaches, dizziness, nosebleeds, leg pain, and weak or absent femoral pulses.
- Pulmonary Stenosis (PS): Narrowing of the pulmonary valve.
- Cyanotic Defects (Decreased Pulmonary Blood Flow): These defects result in inadequate oxygenation of the blood, leading to cyanosis.
- Tetralogy of Fallot (TOF): A complex defect involving VSD, pulmonary stenosis, overriding aorta, and right ventricular hypertrophy.
- Clinical Features: Cyanosis (especially during crying or feeding), “tet” spells (episodes of severe cyanosis and hypoxia), systolic murmur, and clubbing of the fingers and toes.
- Transposition of the Great Arteries (TGA): The aorta and pulmonary artery are connected to the wrong ventricles.
- Clinical Features: Severe cyanosis from birth; requires immediate intervention.
- Truncus Arteriosus: A single great artery arises from both ventricles.
- Clinical Features: Severe cyanosis, usually present from birth; requires early intervention.
- Total Anomalous Pulmonary Venous Return (TAPVR): The pulmonary veins connect to the right atrium instead of the left atrium.
- Clinical Features: Cyanosis, often severe; may present with heart failure.
- Tetralogy of Fallot (TOF): A complex defect involving VSD, pulmonary stenosis, overriding aorta, and right ventricular hypertrophy.
Management (General Principles)
Aims:
- Improve oxygen saturation: Ensure adequate oxygen delivery to the tissues.
- Reduce pulmonary vascular resistance: Prevent damage to the lungs.
- Improve cardiac output: Ensure sufficient blood flow to the body.
- Prevent complications: Minimize the risk of heart failure, endocarditis, and other potential problems.
- Maximize long-term survival and quality of life: Achieve optimal growth and development.
Initial Management (Maternity Centre/Primary Care)
- Assessment of vital signs: Continuous monitoring of heart rate, respiratory rate, oxygen saturation, and blood pressure.
- Oxygen therapy: Supplemental oxygen to improve oxygen saturation.
- Fluid management: Intravenous fluids to maintain hydration and blood volume.
- Early identification of severe defects: Prompt recognition of cyanosis or severe respiratory distress necessitates immediate referral.
- Referral: Immediate referral to a tertiary care center with pediatric cardiology and cardiac surgery services.
Hospital Management
- Diagnosis: Detailed evaluation with ECG, chest X-ray, and echocardiogram. Cardiac catheterization may be needed in certain cases.
- Surgical management: Many CHDs require surgical intervention to correct the defect(s). Examples include:
- Cardiac repair: Surgical closure of VSDs, ASDs, or PDA.
- Valvuloplasty/Valve replacement: Procedures to repair or replace narrowed or damaged valves.
- Arterial switch: Surgical correction of TGA.
- Palliative procedures: Procedures to improve blood flow and oxygen saturation temporarily while awaiting definitive surgery.
- Medical management: Medical therapy is often used in conjunction with surgery or alone for milder defects. This may include:
- Diuretics: Reduce fluid buildup.
- ACE inhibitors: Improve cardiac output and reduce pulmonary vascular resistance.
- Digoxin: Improves heart contractility.
- Prostaglandin E1: Keeps the ductus arteriosus open in certain cyanotic defects.
Nursing Management
- Monitoring vital signs: Continuous monitoring of heart rate, respiratory rate, oxygen saturation, blood pressure, and urine output.
- Medication administration: Accurate administration of prescribed medications.
- Fluid management: Careful monitoring of intravenous fluids and electrolyte levels.
- Oxygen therapy: Administering oxygen as prescribed and monitoring oxygen saturation.
- Pain management: Providing comfort measures and administering analgesics as needed.
- Nutritional support: Assisting with feeding and nutritional support.
- Education and support: Providing education and emotional support to parents.
CNS (Central Nervous System) Abnormalities
Hydrocephalus
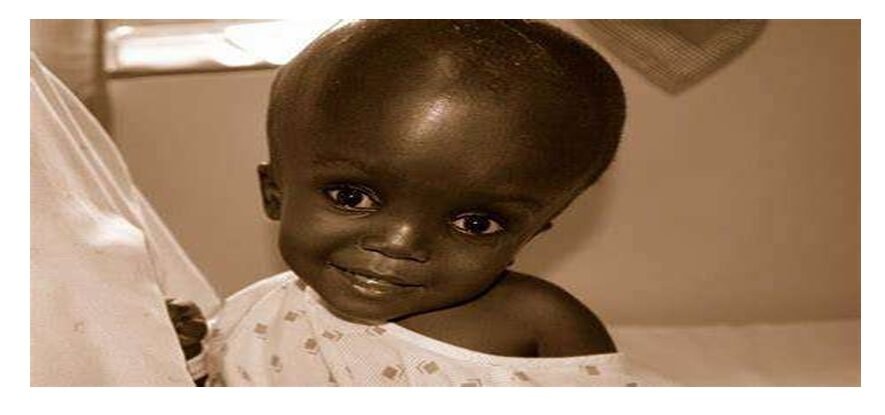
Hydrocephalus is a condition where there’s an abnormal buildup of cerebrospinal fluid (CSF) in the brain.
This fluid normally cushions the brain and spinal cord, but when it can’t drain properly, it puts pressure on the brain. This can cause the head to swell and lead to symptoms like headaches, nausea, vomiting, and changes in vision. In babies, whose skulls are still forming, it can cause the head to enlarge rapidly.
Convulsions in Infants and Young Children
Convulsions, also known as seizures, are involuntary spasmodic contractions of the skeletal muscles due to abnormal electrical activity in the brain.
They represent a malfunction of the brain’s electrical system and are a common neurological presentation in children, particularly during the first two years of life.
The underlying causes are diverse and necessitate a thorough investigation.
Musculo-Skeletal Abnormalities
Amelia and Ectromelia
- Amelia: The complete absence of one or more limbs. Thalidomide exposure during pregnancy is a known cause, but other genetic and environmental factors can also contribute.
- Ectromelia: The partial absence of one or more limbs, a more common occurrence than amelia. The severity can vary widely, ranging from minor limb shortening to significant malformations. Etiology is often multifactorial and may involve genetic factors, teratogens, and vascular disruptions during development.
Polydactyly (Extra Digits)
The presence of more than the usual number of fingers or toes. Diagnosis is typically straightforward through visual inspection. Management depends on the digit’s structure:
- Skin-only attachment: These extra digits can often be simply ligated (tied off) with a sterile thread at birth.
- Digit with bone and firm attachment: Surgical removal is usually necessary and should be performed by a surgeon specializing in pediatric orthopedics or hand surgery. The timing of surgery is determined by the surgeon based on the child’s age and development.
Syndactyly (Webbing of Digits)
Fusion of two or more fingers or toes. The degree of fusion varies. Surgical separation is typically performed to improve function and aesthetics, particularly if the digits are naturally separated but connected by skin or soft tissue. The timing of surgery is determined by the surgeon based on the child’s age and development.
Talipes (Clubfoot)
An abnormal positioning of the foot, often involving inversion (inward bending) and plantarflexion (pointing downwards). It’s frequently caused by intrauterine pressure, such as in multiple pregnancies (twins, triplets) or pregnancies with oligohydramnios (reduced amniotic fluid). Muscle tendon contractures contribute to the deformity.
- Talipes Equinovarus: The most common type, characterized by inward bending (inversion) of the foot and plantarflexion at the ankle.
- Talipes Calcaneovalgus: The foot is turned outward (eversion) with dorsiflexion (upward bending) of the ankle.
Achondroplasia (Chondrodystrophy)
A genetic disorder affecting bone growth, inherited as an autosomal dominant trait. It primarily disrupts cartilage production, particularly in the epiphyseal plates (growth plates) of long bones.
Congenital Dislocation of the Hip (CDH)
The hip joint is dislocated at birth. The cause is often laxity (looseness) of the hip joint capsule, potentially influenced by hormonal factors during pregnancy (e.g., progesterone).
Join Our WhatsApp Groups!
Are you a nursing or midwifery student looking for a space to connect, ask questions, share notes, and learn from peers?
Join our WhatsApp discussion groups today!
Join NowWe are a supportive platform dedicated to empowering student nurses and midwives through quality educational resources, career guidance, and a vibrant community. Join us to connect, learn, and grow in your healthcare journey
Quick Links
Our Courses
Legal / Policies
Get in Touch
(+256) 790 036 252
(+256) 748 324 644
Info@nursesonlinediscussion.com
Kampala ,Uganda
© 2026 Nurses online discussion. All Rights Reserved